
Rethinking UMI Errors in the Era of High-Resolution Transcriptomics
Types, Methods, Innovations + (as always) latest News including M&A

Today we have a guest post by Jianfeng Sun, who has been drilling into the nitty gritty of UMIs, how they work, and how we can do better! As usual, we’ve put a few market updates at the end that caught our attention.
As single-cell RNA-seq (scRNA-seq) continues to scale toward tens of millions of cells per dataset, Unique Molecular Identifiers (UMIs) remain the backbone of accurate molecular counting. Yet, these molecular barcodes, which were originally introduced as an elegant fix to PCR duplication, are increasingly recognised as a source of noise themselves. From synthesis inaccuracies to sequencing errors, UMIs bring their own set of challenges that can no longer be ignored.
What are UMIs?
UMIs are a type of short molecular barcodes of approximately 8–12nt in size. During library preparation, UMIs are ligated to the 3′ or 5′ ends of DNA or cDNA fragments (i.e., original templates). These UMIs are co-duplicated with the fragments during PCR amplification and subsequently sequenced along with them. After sequencing, reads sharing the same UMI are collapsed into one, thereby removing PCR duplicates (Figure 1). However, synthesis errors within UMIs can prevent correct merging of reads derived from the same original template, usually leading to inflated gene expression estimates.

Figure 1. Location of the UMI in a read using bulk RNA-seq or single-cell RNA-seq (scRNA-seq).
For molecular quantification, errors within the UMI regions need to be corrected rather than those on the DNA fragments. To achieve this, both bioinformatic and experimental approaches have been conceived.
Why is UMI error correction challenging?
UMI sequences are typically synthesised in a completely random manner and lack any traceable origin or predefined whitelist. As a result, it is inherently unknown which UMI is attached to which DNA fragment, or even how many distinct UMIs exist in a given dataset. This randomness introduces significant challenges for mathematical modelling and computational analysis, particularly in accurately identifying and correcting errors or estimating molecular counts (Figure 2).
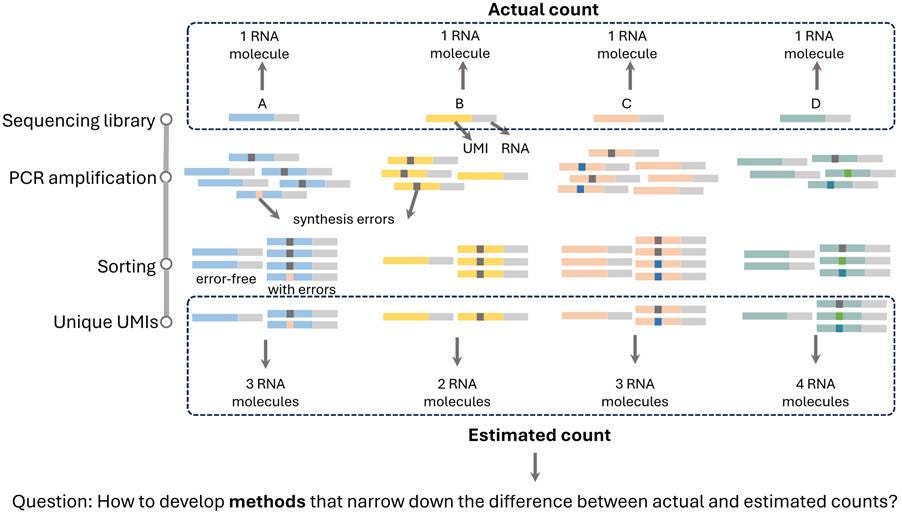
Figure 2. Illustration of error-aware molecular quantification using UMIs. During library preparation, each RNA molecule (A–D) is uniquely labelled with a UMI. PCR amplification then produces multiple copies of each molecule with synthesis errors in UMI sequences. As a result, sequencing yields a mixture of error-free and erroneous UMI-tagged reads. Without error correction, these reads can lead to incorrect UMI counting, generally inflating the estimated number of unique molecules. For example, a single original RNA molecule may be mistakenly counted as 2 or 3 distinct molecules. The challenge in UMI-based molecular quantification underscores the need for methods capable of effectively correcting UMI errors for precise count estimates.
Categories of UMI errors
UMI errors arise from three major sources: PCR errors, sequencing errors, and bead truncation errors (Figure 3). These can introduce subtle distortions into molecular quantification, especially under conditions of deep amplification, long-read sequencing, or imperfect probe manufacturing. In many datasets, particularly those relying on ultra-sensitive protocols or barcoded bead libraries, these “small” issues are no longer negligible. For example, our research demonstrates that these errors can cause more than 25% of genes identified as differentially expressed to be false positives.
PCR amplification errors
PCR amplification introduces random nucleotide substitutions that accumulate over multiple cycles. As each round of amplification uses previously synthesised products as templates, even low-frequency errors can propagate and become fixed in downstream reads. This compounding effect leads to an increased likelihood of erroneous UMIs being interpreted as distinct molecules. The error rate significantly increases with each PCR cycle, making it especially problematic in contexts like single-cell sequencing, where limited input material necessitates extensive amplification to generate sufficient DNA for library preparation and sequencing. Consequently, without proper error correction, these accumulated UMI errors can distort molecular counts and compromise the accuracy of downstream analyses.
Sequencing errors
Sequencing errors refer to incorrect base calls that occur during the sequencing process, leading to mismatches between the readout and the original DNA or RNA template. These errors can be nucleotide substitutions, insertions, or deletions, and are an inherent limitation on all sequencing platforms. The nature and frequency of these errors vary widely depending on the underlying sequencing technology. For example, Illumina platforms, which rely on sequencing-by-synthesis, typically exhibit low overall error rates but are mainly affected by substitution errors. In contrast, long-read platforms such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) are more susceptible to insertion and deletion errors due to their real-time sequencing chemistry that works with single molecules. Understanding the error profiles of different platforms is essential for designing effective UMI error correction strategies, particularly when integrating data from multiple sequencing sources.
Oligonucleotide synthesis errors
Oligonucleotide synthesis errors occur during the chemical manufacturing process of UMIs, primarily involving truncation (premature termination) and elongation (unintended extensions). Chemical synthesis typically employs solid-phase phosphoramidite methods, which inherently have finite efficiency. Each synthesis step has a coupling efficiency of approximately 98-99%, meaning a small percentage of sequences fail to extend correctly at each step. Consequently, as the length of UMIs increases, the cumulative probability of synthesis errors rises, causing a large proportion of oligonucleotides to deviate from their intended sequences. These deviations can introduce substantial biases in molecular counting, reducing the reliability of downstream analyses and potentially compromising biological interpretations.
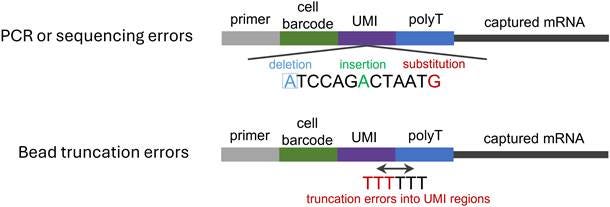
Figure 3. Schematic of error types over the whole sequencing process. Errors introduced in PCR and sequencing steps include deletions, insertions, and substitutions. In contrast, bead truncation errors detected during library preparation primarily refer to contamination or misreading of the UMI tail by poly(T) sequences.
State of the art for UMI error correction
Recently, R&D in UMI error correction is accelerating rapidly, driven by a joint effort between experimental and computational innovations. While some computational tools like UMI-tools and mclUMI remain effective for substitution errors in short-read datasets, they struggle with indels and complex UMI settings. To address these limitations, a couple of recent studies have begun to explore ideas of designing new UMI types prior to developing new computational methods, such as homodimer UMIs and homotrimer UMIs. The UMI designs introduce internal redundancy and offer new opportunities for more robust and structure-aware error correction. More adaptive approaches like majority voting and set cover optimisation, leveraging homomultimeric units, have emerged and improved performance under some challenging conditions, such as high-error rates. Rather than relying on this single-stage process for error correction, a recently released platform, UMIche, has reported a two-stage pipeline that integrates data preprocessing, majority voting, and distance-based graph inference, offering an even more pronounced improvement in silico. The new UMI designs and their relevant computational methods are discussed below.
1. UMI structures
A.) Structural innovations by homotrimer UMIs
Homotrimer UMIs represent a structural innovation inspired by the concept of triple modular redundancy (TMR), commonly used in fault-tolerant computing and cryptography. In this design, each nucleotide in a conventional UMI is replaced by a triplet of identical bases (e.g., A becomes AAA, G becomes GGG), forming repeated blocks that increase the sequence length while introducing redundancy.
This redundancy enables majority voting within each triplet to correct single-base substitution errors during sequencing or amplification. For example, if a true AAA triplet becomes ATA due to an error, the system can still infer the correct base (say A) because it appears more than once. This mechanism significantly improves the robustness of UMI decoding, especially under high PCR cycle conditions where error rates tend to increase.
Importantly, even in the worst-case scenario where all three bases in a triplet differ (e.g., AGC) and majority voting cannot resolve the correct base. However, the design still retains a chance of correct interpretation through random selection or set-cover-optimisation-based strategies, such as choosing the most frequently observed decoded UMI across reads. These fallback mechanisms, while not perfect, still provide better reliability than standard monomer UMIs, which lack internal redundancy.
Homotrimer UMIs have demonstrated superior performance across multiple sequencing platforms, offering a biologically relevant adaptation of repetition code principles. Their enhanced error tolerance makes them especially useful in settings where accurate molecular counting is critical, such as single-cell transcriptomics.
B.) Anchor oligonucleotide sequence design
Another recent structural innovation for improving UMI accuracy is the introduction of an anchor sequence, a short, predefined oligonucleotide segment inserted between the cell barcode and the UMI region on a sequencing bead. This approach addresses a specific yet prevalent source of error: oligonucleotide synthesis inaccuracies, particularly truncation errors that occur during the manufacturing of bead-bound primers used in high-throughput droplet-based methods like 10x Genomics or Drop-seq.
Due to incomplete synthesis of oligonucleotide sequences, part of the UMI may be missing, misaligned, or incorrectly interpreted by the sequencing pipeline. This leads to misassignment of reads, inaccurate molecular counts, and inflated noise in downstream gene expression analyses.
The anchor sequence acts as a positional landmark, clearly delineating where the barcode ends and the UMI begins. With this fixed reference point, computational pipelines can reliably detect and extract the UMI even when the actual oligonucleotide may be truncated or slightly malformed. In other words, the anchor makes it possible to distinguish a true UMI from synthesis artifacts by providing a stable and recognisable junction. Our studies have shown that this structural adjustment leads to i.) a higher fraction of reads with correctly identifiable UMIs, ii.) reduced bias in base composition at UMI start sites, and iii.) improved consistency in UMI counts across cells and genes.
Importantly, this approach is complementary to error correction strategies aimed at sequencing or PCR errors. While methods like homotrimer UMIs improve fault tolerance post-sequencing, anchor sequence design enhances pre-sequencing fidelity by reducing the chances of incorrect UMI structure from the outset.
2. Computational methods
A.) UMI-tools for monomer UMIs
Graph-based methods, such as those implemented in UMI-tools, have most widely been used to counteract UMI errors in the field of RNA-seq. These methods use edit distances to cluster and merge similar UMIs. While effective in PCR artefact removal against moderate errors, these methods are notably less effective in addressing high-error profiles.
B.) Markov clustering (MCL) methods for monomer UMIs
The mclUMI tool applies a graph-based approach using the Markov cluster algorithm (MCL) to correct UMI errors. Unlike traditional methods that rely on fixed Hamming distance thresholds, mclUMI builds a graph where UMIs are nodes and edges connect similar sequences. The number and size of UMI clusters are controlled by two parameters, expansion and inflation, to adjust how tightly UMIs are grouped. This allows the method to adaptively identify true molecular counts without relying on fixed edit distance thresholds. This method is particularly effective under high-error conditions, such as extensive PCR amplification or sequencing noise, and offers improved accuracy for low-abundance transcripts compared to static clustering methods.
C.) Integrated platform methods
UMIche represents an integrated approach for UMI error correction. Rather than relying on a single strategy, it combines multiple complementary algorithms, including graph-based clustering, distance-based filtering, and set cover optimisation, into a unified platform that systematically addresses a wide range of UMI error types. It has the following features.
Distance-based clustering methods. UMIs are compared using sequence similarity metrics (such as Euclidean distance), and similar sequences are grouped under the assumption that minor differences likely result from errors during sequencing.
Graph-based clustering methods. A graph is constructed where nodes represent UMIs and edges indicate sequence similarity. Connected components or subgraphs are identified as potential groups of UMIs originating from the same molecule.
Set cover optimisation method. To resolve ambiguities in overlapping UMI clusters and minimise redundancy, UMIche applies a set cover algorithm to choose the minimal representative monomer UMI set that explains all observed homotrimer UMIs. This step helps avoid over-collapsing distinct molecules that happen to have similar UMI sequences.
Two-stage UMI error correction. UMIche implements a two-stage error correction framework tailored for homotrimer UMI structures. In the first stage, each 36-bp homotrimer UMI is decomposed into 12 individual monomer units, and a set cover algorithm is applied to identify and eliminate redundant or conflicting homotrimer UMI groups. This step effectively resolves many early-stage errors arising from PCR amplification or sequencing artifacts that inflate molecular counts. In the second stage, monomer UMI deduplication methods, such as graph-based clustering or edit-distance-based merging, are applied to the refined set of homotrimer UMIs to further correct residual substitution errors at the monomer level. This layered strategy enables both structural redundancy correction and fine-grained sequence-level error reduction, which enhances the reliability of molecule-level quantification. In short, UMIche exemplifies how combining computational techniques in an integrated framework can lead to more accurate and robust UMI resolution. It helps ensure reliable molecular counting in high-throughput sequencing data.
Expert advice
The field of UMI error correction has recently been propelled by both structural innovations and computational advancements tailored for the complexities of single-cell sequencing. Recent developments reveal that no single strategy can universally resolve all forms of UMI errors. Instead, an integrated, multi-layered framework, which is attuned to the origin and nature of the errors, delivers the most robust performance. In practical terms, the following is advised:
For scenarios dominated by substitution errors and extensive PCR amplification, homotrimer UMIs are highly recommended.
Anchor sequences might be the design of choice to address oligonucleotide synthesis and truncation errors in sequencing libraries.
Traditional monomer UMIs adequately correct moderate substitution errors but falter significantly under conditions involving high error rates. For comprehensive and dynamic error correction, integrated computational platforms like UMIche are valuable.
Integrating these complementary strategies offers a robust, multi-layered defense against UMI errors, ensuring greater accuracy and reliability in single-cell sequencing analyses.
Market news:
15-18MAY25 - Tough times for 10x
Single-cell pioneer, 10x Genomics is feeling the pinch of the NIH funding cuts, leading to an 8% workforce reduction. This may mark a key transition point for the industry as capital markets want to see a renewed focus on industry-funded studies, when compared to academia. In related news, 10x threw in the towel to settle patent lawsuits with both Vizgen and Bruker.
13MAY25 - Nomic’s $50 proteome
Using a dual antibody system not dissimilar to to O-link, Nomic Bio have released a $50 proteome. As the validation data and technology matures, we may be seeing a meaningful shift downwards in price.
23JUN25 - Illumina to acquire SomaLogic from Standard BioTools
The original merger between SomaLogic and STB (originally Fluidigm) didn’t make the original markets review, but clearly Illumina does not want to sit on the sidelines of the genomics revolution — remember the acquisition of Fluent BioSciences? Special thanks to Alex Dickinson’s phenomenal Linkedin post, his commentary suggests Standard BioTools were only too happy to offload SomaLogic.
 | A guest post by
|